



")

卷 101, 编号 4 (2024)

- 年: 2024
- 文章: 11
- URL: https://journals.rcsi.science/0372-9311/issue/view/16959

ORIGINAL RESEARCHES
Genomic surveillance of SARS-CoV-2 in Russia: insights from the VGARus platform
摘要
Introduction. In response to the COVID-19 pandemic in the Russian Federation, comprehensive response measures were taken. One of these measures was the development of a viral genome aggregation platform (VGARus) to monitor virus variability.
The aim of this paper is to describe the role of the VGARus platform in tracking genetic variation in SARS-CoV-2.
Materials and methods. VGARus utilizes sequencing data and bioinformatics tools to monitor genetic variations in SARS-CoV-2. The viral genomes were aligned using NextClade, which also translated them into amino acids and identified mutations. The viral variability over time was analyzed by counting the number of amino acid changes compared to the reference sequence.
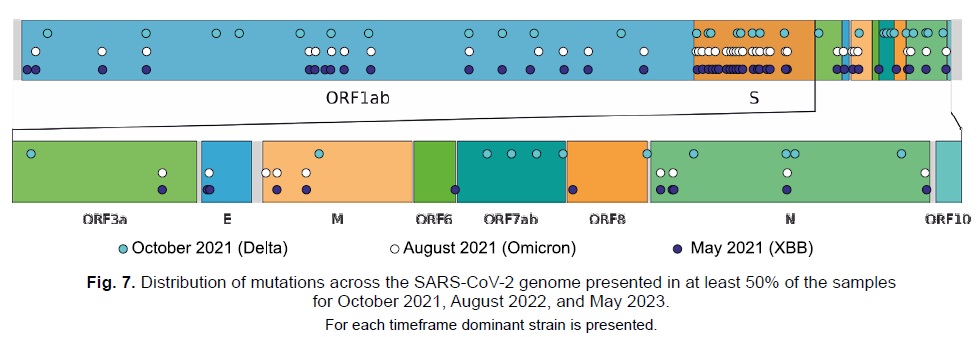
Results. The analysis of data within VGARus enabled the identification of new virus variants, contributing to improved diagnostic tests and vaccine development. The platform allowed for the prediction of epidemiologic trends, facilitating a rapid response to changes in the epidemiologic situation. For example, using VGARus, an increase in COVID-19 incidence was accurately predicted in the summer of 2022 and early 2023, which were associated with the emergence of Omicron subvariants BA.5 and XBB. Data from the platform helps validate the effectiveness of primers and DNA probes to ensure high diagnostic accuracy and reduce the risk of false negatives.
Conclusion. VGARus demonstrates the growing role of genomic surveillance in combating COVID-19 and improving preparedness for future infectious disease outbreaks. The platform is a powerful tool for generating evidence-based solutions to combat a pandemic and mitigate its health, economic and societal impacts. It provides the ability to promptly obtain information on the epidemiologic situation in a particular region of the Russian Federation, use genomic data for phylogenetic analysis, compare the mutational spectrum of SARS-CoV-2 sequences with foreign samples. VGARus data allow for both retrospective analysis and predictive hypotheses. For example, we can clearly see the dynamics of the change of different virus variants: sequences belonging to the Alpha, Beta, Delta, Omicron lineages and many less common ones, clearly form the upsurges of morbidity, the interaction of which is reflected in the epidemiological picture. It is also currently being expanded to monitor other pathogens, increasing its public health relevance.
 435-447
435-447



Anthrax in the Russian Federation in 2023 or in other words, «the same old story»
摘要
Introduction. The current anthrax situation in Russia is characterized by instability. In 2023, there was an increase in the number of infection outbreaks compared to the long-term average (for five years).
The aim of the study is to assess the epizootological and epidemiological situation regarding anthrax in the Russian Federation in 2023 and the reasons for its deterioration, and to analyze data from genomic epidemiological surveillance of this infection.
Materials and methods. The information of the territorial bodies of Rospotrebnadzor on the investigation of anthrax outbreaks, reference materials about anthrax stationary hazardous areas and anthrax burials were used. The phylogenetic position of the identified Bacíllus anthracis strains and genomes structure were determined based on whole-genome sequencing data.
Results. In 2023 anthrax outbreaks were registered in the Chuvash Republic — Chuvashia (1), the Tyva Republic (1), Tambov (1), Ryazan (1) and Voronezh (3) regions. 14 farm animals and 19 people fell ill. The infection of animals not vaccinated against anthrax, as well as vaccinated long before contact with the source of infection, occurred mainly during grazing in the territories of old (unregistered) anthrax soil foci. Human disease is caused by contact with sick animals during care, forced slaughter, cutting, transportation of carcasses and meat, cooking processing of contaminated meat and offal, and consumption of insufficiently heat-treated liver. 17 patients were diagnosed with a cutaneous form of anthrax, while 2 had an oropharyngeal form combined with a cutaneous form of the disease.
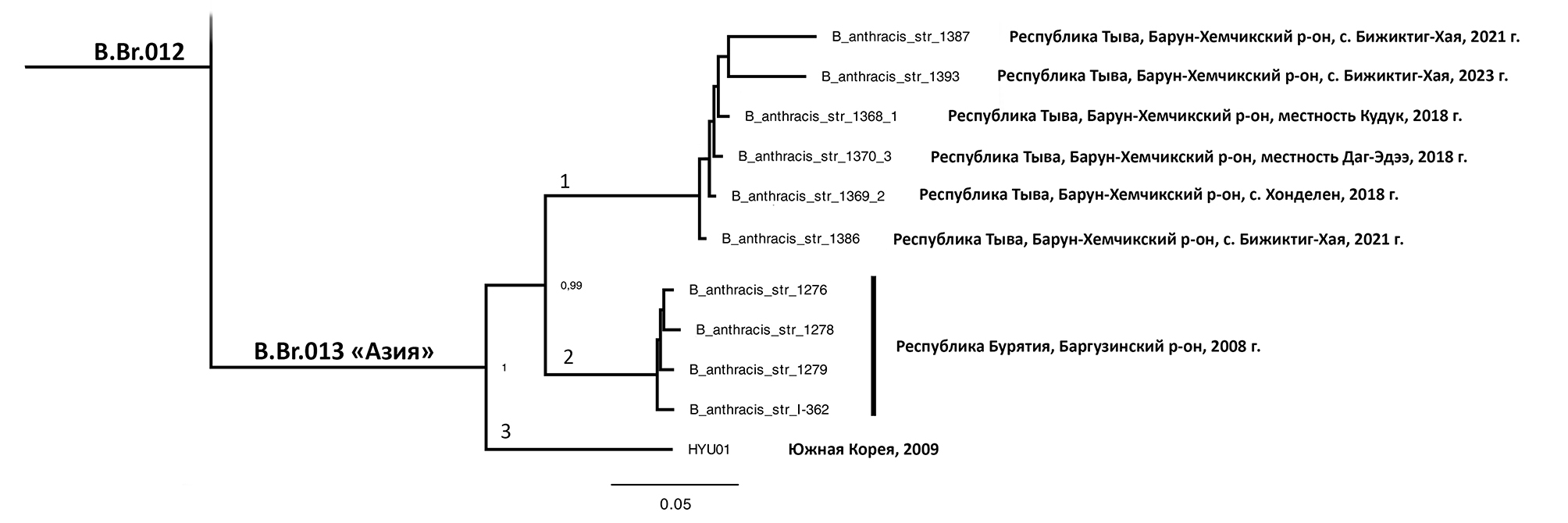
In all cases, the genome structure typical of the B. anthracis species has been established. The phylogenetic relationship of B. anthracis isolates with B. anthracis strains previously isolated in Russia is shown.
Conclusion. The reason for the trouble in anthrax in 2023 was a number of violations of veterinary and sanitary-epidemiological regulations against the background of the presence of soil foci of infection. Stabilization of the situation can be achieved only in full range of regulated preventive measures are constantly implementated. The results of molecular genetic typing of B. anthracis strains isolated during the epidemiologic investigation of seven anthrax outbreaks in the Russian Federation in 2023 allow us to conclude that they are of local origin and have a genome structure typical of the species. Genetic analysis of the isolated strains demonstrated the effectiveness of the developed wgSNP typing system in the epidemiologic investigation of outbreaks.
448-461
The molecular-genetic characteristics of uropathogenic Escherichia coli isolated from pregnant women with asymptomatic bacteriuria
摘要
Introduction. Uropathogenic Escherichia coli (UPEC) are the dominant bacterial pathogens of urinary tract infections (UTIs). UPEC belong to different phylogenetic groups and have many virulence factors, the study of which, in conjuction with the assessment of their relationship with clinical forms of UTI, is necessary for a better understanding of the pathogenesis of UTI and the development of new diagnostic algorithms.
Aim: determination of the molecular genetic characteristics of uropathogenic Escherichia coli isolated from pregnant women with asymptomatic bacteriuria.
Materials and methods. Clinical isolates of uropathogenic E. coli (n = 70) from pregnant women with asymptomatic bacteriuria were included in the study. The PCR method was used to determine the belonging to phylogenetic groups and detect 15 virulence markers — genes associated with adhesion (fimH, papC, sfa, afa, focG); toxin synthesis (cnf 1, hlyA, sat, vat, usp); siderophores (fyuA, iroN, iuc); capsular antigen (kpsMII). To assess the statistical significance of differences, Fisher's exact test was used. Differences were considered statistically significant at a confidence interval of 95% (p < 0.05).
Results. Most of the UPEC isolates belonged to phylogroup B2 (51,4%) and were characterized by the detection of all UPEC-associated virulence factors included in this study; genes associated with adhesion (sfa, focG), invasins (ibeA), synthesis of toxins (hlyA, cnf1, vat, usp) and capsule (kpsMII), siderophores (fyuA, iroN, hlyA) were detected significantly more frequently (p < 0.05). Two or more virulence determinants were detected in 93% of isolates.
Conclusion. The identification of key determinants of virulence and/or a combination of virulence genes can be a prognostic marker for predicting the course of UTI, especially in pregnant women, and will expand diagnostic capabilities taking into account the virulent properties of the uropathogen.
462-469
Virulence and tissue tropism of different epidemiologically significant SARS-CoV-2 variants for golden Syrian hamsters
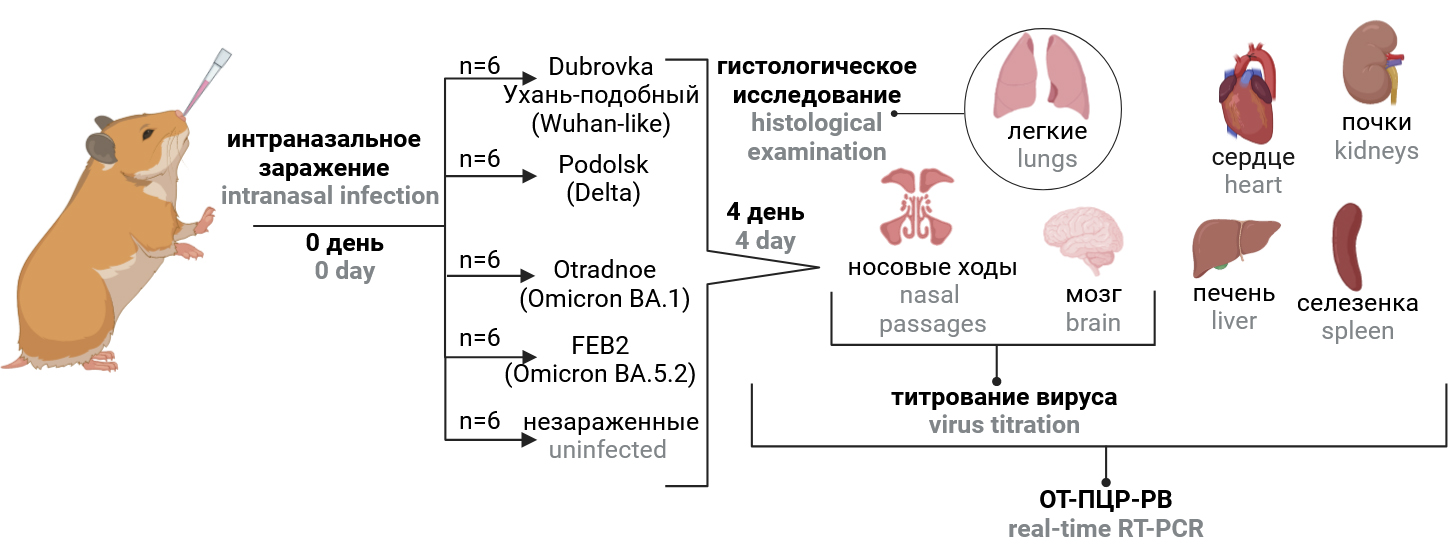
摘要
Introduction. Animal models for SARS-CoV-2 infection, reproducing the clinical features of COVID-19 in humans, are important tools for studying the pathogenesis of the disease, transmission of the pathogen and are indispensable for testing antiviral drugs and vaccines.
The aim of the study was to assess the virulence and tissue tropism for golden Syrian hamsters of SARS-CoV-2 strains belonging to different epidemiologically significant variants: Wuhan-like, Delta, Omicron BA.1.1 and Omicron BA.5.2.
Materials and methods. Hamsters were intranasally infected with different SARS-CoV-2 strains. Virulence and tissue tropism of SARS-CoV-2 strains were assessed by comparing the dynamics of weight, viral load in organs and histopathological changes in lungs in infected and uninfected animals.
Results. The Wuhan-like Dubrovka strain had the greatest virulence for hamsters, which was manifested by the development of severe pneumonia and a delay in weight gain by 14.6%, high virus content in the lungs, nasal passages and brain — 6.2, 5,9 and 3.7 lg TCID50/ml of homogenate, respectively. Presumably, it was the infection of the Wuhan-like virus of the central nervous system that negatively affected the weight and general condition of the animals. When hamsters were infected with viruses belonging to the Delta and Omicron variants, the observed minor weight loss in animals was uninformative, so indicators such as lung histopathology, viral load in the lungs, nasal passages, heart and other organs played a decisive role in assessing the virus pathogenicity. A score assessment of lung histopathology was of particular value in assessing the severity of pneumonia, since it reduced subjectivity in evaluating the results of histological examination and provided a semi-quantitative assessment of the pathological process.
Conclusion. Despite the revealed lower virulence for hamsters of viruses belonging to the Delta and Omicron variants compared to the ancestral Wuhan virus, this animal model for COVID-19 retains its value for conducting preclinical trials of antiviral drugs.
470-482
Molecular genetic characteristics of Streptococcus pneumoniae serogroups 15 and 11 representatives circulating in Russia and their relationship with global genetic lineages
摘要
Aim of the study. Genetic analysis of Streptococcus pneumoniae serogroups 15 and 11 circulating in Russia according to the following parameters: serotype affiliation; clonal complex (CC); presence of resistance and virulence determinants; relatedness to genetic lineages circulating in the world, and justification of inclusion of the actual serotypes of serogroups 15 and 11 in the future conjugate vaccine composition.
Materials and methods. The study included whole genome data of S. pneumoniae serogroups 11 and 15.
Results. Genomes of serogroup 15 strains from Russia are represented mainly by serotypes 15B and 15C, the majority of which belong to CC-1025 and CC-1262. CC-1025 is characterized by a more frequent association with invasive diseases. Representatives of CC-1025 and CC-1262 contain virulence determinants unique to these genetic lineages within the studied population of serogroup 15: oligopeptide transporters, fructose-specific PTS system, unique hydrolase variants, additional iron ion transporters, the gene of zinc metalloprotease ZmpC (activating human MMP9). The genomes of serogroup 11 are represented mainly by serotype 11A, the majority belong to CC-62 and CC-1012. The virulence determinants unique to CC-62 (within the studied serogroup 11) include bacteriocins, components of oligopeptide transport, flavin reductase-like protein (adhesin, also protects bacteria from oxidative stress), fucose processing operon, PsaA (adhesin, also a component of the ATP-binding cassette transporter that imports manganese ions).
Conclusion. In the Russian Federation, serogroups 15 and 11 are the most common non-vaccine serogroups. No antimicrobial resistance determinants have been identified in the genomes of representatives of these serogroups. For each of the genetic lineages prevalent in Russia and associated with serogroups 15 and 11, unique virulence determinants within the studied serogroup have been identified, which may contribute to the success of these lineages. It is advisable to include serotypes 15B and 11A in vaccines promising for the Russian Federation.
483-501
Prevalence of qacEΔ1, qacE, oqxA, oqxB, acrA, cepA and zitB genes among multidrug-resistant Klebsiella pneumoniae isolated in a cardiac hospital
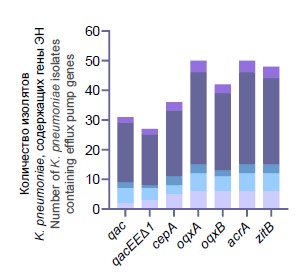
摘要
Background. Infections caused by multidrug-resistant (MDR) Klebsiella pneumoniae are the leading cause of mortality worldwide. The widespread use of disinfectants and antiseptics has caused the emergence of K. pneumoniae with reduced sensitivity to them, which, in combination with MDR, can pose a significant epidemiological threat.
The aim of the study was to assess the prevalence of efflux pump and transporter genes associated with biocide resistance and their association with antibiotic resistance among K. pneumoniae isolated in a cardiac surgical hospital.
Materials and methods. K. pneumoniae isolates (n = 50) from the patients and medical equipment were tested by polymerase chain reaction for the presence of genes of 4 types of efflux pumps (qacEΔ1, qacE, oqxA, oqxB, acrA) and 2 transporters involved in the outflow of cations (cepA) and zinc ions (zitB). Spearman's rank correlation test was used to assess the strength of the association between the efflux pumps, beta-lactamase genes and mobile genetic elements.
Results. The occurrence of K. pneumoniae containing qacEΔ1, qacE, oqxA, oqxB, acrA, cepA and zitB was high: 54, 62, 100, 84, 100, 72 и 96% respectively. K. pneumoniae with a combination of all the studied pumps was most often detected (32%), and these isolates were MDR in 100% of cases. The qacE, qacEΔ1 genes were closely associated with resistance to cephalosporins, carbapenems, fluoroquinolones, carbapenemase genes, and integrons. The results of the study showed that the genes of various efflux pumps associated with biocide resistance and their combinations were widely represented among the clinical isolates of MDR K. pneumoniae.
Conclusion. The high prevalence of efflux pump genes associated with resistance to quaternary ammonium compounds, chlorhexidine and zinc salts and their significant association with antibiotic resistance in nosocomial K. pneumoniae underlines the importance of further studying the mechanisms of cross-resistance to biocides to improve methods of combating MDR nosocomial pathogens.
502-511
Evaluation of symbiotic relationships of oral microorganisms and their effect on the development of inflammatory changes of the oral mucosa in the complete absence of teeth
摘要
Introduction. By fixing on the exposed surfaces of complete removable dentures and oral soft tissues, bacteria form a biofilm, thereby increasing their overall virulence and resistance. The microorganisms that make up the biofilm are often in a symbiotic relationship, which allows them to increase their pathogenic potential and cause the development of denture stomatitis. Accordingly, when a particular strain is present in the oral cavity, the risks of symbiosis are significantly increased.
The aim of the study was to evaluate the effect of symbiotic relationships of oral bacteria on the development of inflammatory changes in the oral cavity in the absence of teeth.
Materials and methods. Two groups of patients belonging to the elderly age according to WHO systematization (60–74 years) with complete absence of teeth (K08.1) were formed, differing in the presence of clinical manifestations of inflammation (82 men and 49 women). Biological material sampled from the oral cavity of patients was studied using the culture method and RT-PCR. To quantify the interaction between members of the microbiocenosis, we used the Jaccard similarity coefficient.
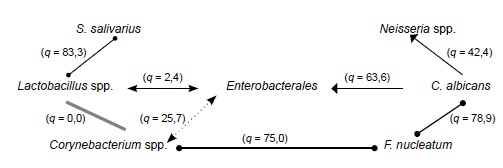
Results. Coagulase-negative and coagulase-positive staphylococci, Neisseria, Candida spp., Enterobacterales and F. nucleatum were more frequently found in patients with complete absence of teeth. Expressed symbiotic relations between microorganisms of the Enterobacterales order, Lactobacillus, Neisseria and Corynebacterium genera, as well as S. salivarius, C. albicans, F. nucleatum were established. The nature of these relations depended on the presence of inflammatory changes in the oral mucosa and, in turn, influenced the development of the latter. Thus, in the absence of inflammation, Corynebacterium, Lactobacillus and S. salivarius showed stable synergism. In case of inflammation, the association between these bacteria was accompanied by the introduction of F. nucleatum and displacement of S. salivarius.
Conclusion. Thus, conditionally pathogenic microorganisms, forming microbial associations with multidirectional symbiotic relations increase their virulence, which allows them to occupy free niches in the oral cavity and subsequently trigger the development of pathological process of inflammatory character of prosthetic bed tissues.
512-519
Comparative analysis of the structure of regulatory genes of Vibrio cholerae serotype О1 biotype El Tor strains
摘要
Introduction. The expression of the ctxAB and tcpA-F genes encoding the main pathogenicity factors of the Vibrio cholerae is controlled by regulatory genes. The structure of these genes has not been fully studied in the pathogen strains isolated during different periods of the current pandemic.
The aim of the study was a comparative analysis of the structure of regulatory genes of V. cholerae O1 biovar El Tor strains isolated on the territory of the Russian Federation and neighboring countries during the seventh cholera pandemic.
Materials and methods. The nucleotide sequences of the complete genomes of 29 toxigenic strains isolated from 1970 to 2023 were analyzed. The analysis was carried out using BioEdit v7.2.6.1 software and Blast tool.
Results. The analysis of ten regulatory genes (toxT, aphA, aphB, hns, hapR, vieA, luxO, luxT, carS, carR) was carried out. Almost all strains were found to have a thymine insertion in the hapR gene at position 219. The exception was V. cholerae strain M3208 (Tambov, 2023), which had an insertion of five nucleotides in this gene. Mutations of the luxO gene with an unknown effect were detected in 44.8% of the studied strains. In 46.7% and 33.3% of the studied genetic variants carrying the ctxB1 allele, non-synonymous substitutions were detected in the hns (G319A) and vieA (C235T) genes, respectively. All genetic variants with the ctxB7 allele have mutations in both the hns and vieA genes. Three genetic variants with the ctxB7 allele, imported to the Russian Federation in recent years, contain an altered structure of the carR gene (G265A).
Conclusion. The structure of genes (toxT, aphA, aphB, carS, luxT, hapR) of V. cholerae O1 El Tor strains remains unchanged for the majority of the studied isolates. At the same time, variability in the hns (G319A), vieA (C235T) and carR (G256A) genes was detected. Mutations in these genes can be used as genetic markers of modern V. cholerae O1 El Tor genetic variants.
520-529
REVIEWS
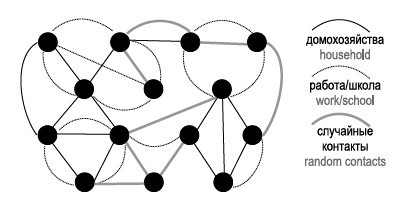
Limitations in creating artificial populations in agent-based epidemic modeling: a systematic review
摘要
Introduction. The key step in agent-based modeling of epidemics, which allows researchers to take into account individual characteristics of people, is the creation of an artificial population. The main difficulty of this procedure is finding a balance between the detail of the population description and the computational efficiency of the calculations.
The aim and objectives of the review: Critically analyze and summarize the current evidence on how to create artificial populations; evaluate the limitations and advantages of available approaches in solving various problems in epidemiology.
Materials and methods. An analysis of literature sources devoted to agent-based modeling has been performed. The analysis is focused on algorithms for creating an artificial population with a given level of detail for modeling human respiratory infections.
Results. The approaches to the creation of artificial populations are generalized. The main principles of realization of interaction between agents are revealed: by means of networks of contacts between agents and on the basis of taking into account the movement of agents between locations. The first approach is the most computationally efficient and simple; the second approach allows to better take into account the change in the behavior of agents during the development of the epidemic process.
Conclusion. Agent-based modeling is an optimal tool for selecting the best scenario for epidemic control and investigating the role of individual characteristics of people in the development of epidemics. When creating an artificial population, it is important to include in the model factors that can be targeted for control. A significant limitation is the lack of factual data on population structure, but this can be overcome by using indirect data.
530-545
The importance of the role of T-cell immunity in the development of modern tick-borne encephalitis vaccines
摘要
The tick-borne encephalitis (TBE) virus is highly pathogenic and can affect the central nervous system, leading to severe chronic effects or death. The only effective measure to combat TBE is vaccine prophylaxis. Vaccines that are currently used for mass immunization are based on inactivated TBE virus, they provide a protective immune response, but such vaccines require multiple administrations. A possible reason for short-term immunity is an incomplete functional T-cell response to these types of vaccines.
The aim of this review is to analyze the literature on the role of the T-cell immune response in protecting the body from tick-borne encephalitis, its importance for vaccine development, and to consider approaches to the development of new TBE vaccines based on different platforms.
When preparing the review, we analyzed the literature presented in scientific databases — PubMed, Scopus, Elsevier, Google Scholar as of April 2024. The following keywords were used for the search: vaccine, tick-borne encephalitis virus, T-cell immune response, flaviviruses.
A several publications have demonstrated that T-cell responses following natural infection with TBE virus and after vaccination with inactivated virus are different. During viral infection, both Th1- and Th2-type CD4+ T cells and CD8+ T cells are activated and play an important role in the elimination of viral infection. After vaccination, the only Th2-type CD4+ T-cell response predominates, which may be the reason for the short-lived immune response.
To date, a number of different types of experimental TBE vaccines are being studied, such as live-attenuated vaccines, viral vector vaccines, subunit vaccines, virus-like particles, DNA and mRNA vaccines, and polyepitope immunogens. In our opinion, the most promising in terms of T-cell response activation are vaccines based on T-cell polyepitope immunogens delivered in the form of DNA or mRNA.
546-559
Assessment of the current state of pharmaceutical development of anti-staphylococcal prophylactic drugs
摘要
Infection caused by Staphylococcus aureus is the most common infection leading to the development of serious complications in humans. S. aureus is among the highly lethal bacteremia-associated pathogens with a mortality rate of approximately 18% in industrial countries; in developing countries, the rate is even higher, up to 27%.
One of the most striking and challenging aspects of clinical manifestations caused by S. aureus is the ability of the bacterium to develop resistance to antibiotics. The development of alternative treatment options for staphylococcal infection is urgently needed. The use of immunotherapy and immunoprophylaxis to activate the anti-infection immune response in patients should be considered as a promising direction.
Objective: to analyze the main trends in the development of vaccines aimed at the prevention of S. aureus infection and its virulence factors.
The present review discusses vaccine development in recent years aimed at preventing infection caused by S. aureus. Particular attention is paid to pathogenicity factors (such as capsule, surface proteins and enzymes) that may be useful for the development of new candidate vaccines or immune therapeutics. In recent years, numerous clinical trials of candidate vaccines based on different antigens, taking into account particularly relevant S. aureus pathogenicity factors that influence morbidity, have not been successful due to their low efficacy or insufficiently substantiated safety (development of adverse events). One of the most important factors constraining vaccine development is the lack of successful translation of vaccine protective activity, which is observed in preclinical studies in experimental models but not confirmed in clinical trials.
Therefore, according to numerous researchers, the use of multiple antigens in vaccine formulations should be considered with the focus on different mechanisms of S. aureus pathogenicity and the use of adjuvants.
560-572