



")

Геномный надзор за SARS-CoV-2 в Российской Федерации: возможности платформы VGARus
- Авторы: Котов И.А.1,2, Аглетдинов М.Р.1,2, Роев Г.В.1,2, Пимкина Е.В.1, Надтока М.И.1, Пересадина А.В.1, Бухарина А.Ю.1, Светличный Д.В.1, Гончаров С.Е.1, Выходцева А.В.1, Борисова Н.И.1, Лысенков В.Г.1, Чанышев М.Д.1, Агабалаев Д.Н.1, Саенко В.В.1, Черкашина А.С.1, Семененко Т.А.3, Дубоделов Д.В.1, Хафизов К.Ф.1, Акимкин В.Г.1
-
Учреждения:
- Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
- Московский физико-технический институт (национальный исследовательский университет)
- Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
- Выпуск: Том 101, № 4 (2024)
- Страницы: 435-447
- Раздел: ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
- URL: https://journals.rcsi.science/0372-9311/article/view/264244
- DOI: https://doi.org/10.36233/0372-9311-554
- EDN: https://elibrary.ru/irjxcx
- ID: 264244
Цитировать

Аннотация
Введение. В ответ на пандемию COVID-19 в России были приняты комплексные меры реагирования. Одной из них стала разработка платформы агрегации вирусных геномов (VGARus) для мониторинга изменчивости вируса.
Цель работы — описать роль VGARus в отслеживании генетических изменений SARS-CoV-2.
Материалы и методы. Выравнивание вирусных геномов, последующую трансляцию в аминокислоты и поиск мутаций производили с помощью программы NextClade. С целью анализа геномной изменчивости подсчитывали число аминокислотных изменений относительно референсной последовательности.
Результаты. Анализ данных VGARus позволил идентифицировать новые варианты вируса, что способствовало улучшению диагностических тестов и может помочь в разработке вакцин. Платформа предоставила возможность прогнозировать эпидемиологические тенденции и оперативно реагировать на изменения эпидемиологической ситуации. Например, с использованием VGARus был точно предсказан рост заболеваемости COVID-19 летом 2022 г. и в начале 2023 г., связанный с появлением субвариантов Omicron BA.5 и XBB. Данные платформы помогают проверять эффективность праймеров и ДНК-зондов, что обеспечивает высокую точность диагностики и снижает риск ложноотрицательных результатов.
Заключение. VGARus демонстрирует растущую роль геномного эпиднадзора в борьбе с COVID-19 и повышение готовности к будущим вспышкам инфекционных заболеваний. Платформа является мощным инструментом для формирования научно обоснованных решений по борьбе с пандемией и смягчению её последствий для здоровья населения, экономики и общества. Она предоставляет возможность оперативно получать информацию об эпидемиологической обстановке в конкретном регионе России, использовать геномные данные для проведения филогенетического анализа, сравнивать мутационный спектр последовательностей SARS-CoV-2 с зарубежными образцами. Данные VGARus позволяют проводить ретроспективный анализ и выдвигать гипотезы прогностического характера. Так, явно можно увидеть динамику смены различных вариантов вируса: последовательности, принадлежащие линиям Alpha, Beta, Delta, Omicron и многим менее распространённым, отчётливо формируют подъёмы заболеваемости, которые отражаются на эпидемиологической ситуации. В данный момент платформа расширяется для мониторинга изменчивости других патогенов, что увеличивает её значимость для общественного здравоохранения.
Полный текст
Введение
Благодаря развитию технологий высокопроизводительного секвенирования, известных как секвенирование нового поколения (next-generation sequencing, NGS), стоимость экспериментов, связанных с определением геномных последовательностей, за последние 15 лет значительно снизилась. NGS всё чаще используется в различных областях биологии и медицины, в том числе в вирусологии, где применение подобных методик для изучения вирусных геномов стало обычной практикой 1–5]. Кроме того, современные средства биоинформатики открыли широкие возможности для создания и анализа баз данных геномов патогенов, вызывающих различные инфекционные заболевания [6–8]. Геномная эпидемиология стала важным средством в борьбе с эпидемиями. Она позволяет изучать генетические изменения, происходящие в геномах патогенов, идентифицировать и классифицировать различные линии, проводить оценку их патогенного потенциала и трансмиссивности [9–15]. Такие исследования очень важны для разработки новых диагностических наборов, современных эффективных вакцин, определения наилучших стратегий противодействия эпидемиям и прогнозирования заболеваемости.
Ярким примером применения молекулярно-генетического мониторинга является детальное изучение новой коронавирусной инфекции во время пандемии COVID-19 [16]. При анализе геномов SARS-CoV-2 были установлены ассоциации между различными вариантами вируса и характеристиками течения эпидемии. Такой подход позволяет точно отслеживать ситуацию, понимать взаимосвязь между генетическими вариантами и их способностью вызывать заболевание, а также внедрять целенаправленные меры для предотвращения распространения инфекции.
В начале пандемии COVID-19 профессор Эдвард Холмс из Университета Сиднея, представляющий команду под руководством Юн-Чжэн Чжана из Фуданьского университета в Шанхае, опубликовал нуклеотидную последовательность вирусного генома. Эта информация была размещена на платформе Virological.org1, что позволило международному научному сообществу начать незамедлительно принимать меры по противодействию распространению патогена, среди которых были разработка новых диагностических тестов и последующее создание вакцин [17, 18]. По мере развития пандемии страны, которые обычно в меньшей степени полагались на собственные геномные данные, начали проводить обширные эксперименты по секвенированию. Полученные знания использовались для разработки стратегических планов, направленных на сдерживание распространения инфекции [19]. Широкое использование секвенирования геномов SARS-CoV-2 привело к значительному увеличению числа новых последовательностей, загруженных в международные базы данных. Самой известной базой стала GISAID (https://www.gisaid.org) с более чем 16 млн последовательностей из более чем 200 стран [8].
Цель исследования — описать роль платформы VGARus и её данных для анализа геномных последовательностей вируса SARS-CoV-2, полученных в России.
Материалы и методы
Перед началом данного исследования было получено информированное согласие пациентов, протокол был одобрен этическим комитетом Центрального научно-исследовательского института эпидемиологии Роспотребнадзора (ЦНИИЭ); протокол № 111 от 22.12.2020. Биологический материал был получен путём взятия мазков из носоглотки у пациентов с симптомами COVID-19. Образцы были собраны из различных регионов России, причём бóльшая их часть поступила из Москвы и Московской области. Наличие РНК SARS-CoV-2 подтверждалось с помощью полимеразной цепной реакции (ПЦР) с обратной транскрипцией в реальном времени. Для выделения РНК использовали набор «RIBO-prep» («АмплиСенс»), для обратной транскрипции — набор реагентов «REVERTA-L» («АмплиСенс»).
Секвенирование выполняли на платформах «Illumina MiSeq» («Illumina») с использованием набора реагентов «MiSeq v2» (PE 150 + 150 или PE 250 + 250 циклов) или «MiSeq v3» (PE 300 + 300 циклов), а также «Illumina NextSeq 2000» с использованием набора реагентов «NextSeq 1000/2000 P2 v3» (300 циклов), «MinION» с использованием набора «Midnight Kit» («Oxford Nanopore Technologies»), «DNBSEQ-G50» с использованием набора «ATOPlex RNA Library Prep Set» («MGI Tech»). Метод Сэнгера использовался для секвенирования фрагментов гена спайкового белка, но эта информация практически не была задействована в анализе. Кроме того, использовались данные нуклеотидных последовательностей из базы данных GISAID в случае их географической принадлежности к России. Программу «Pangolin» [19], а также внутренние инструменты и скрипты применяли для классификации различных вариантов SARS-CoV-2.
Всего использовали более 82 000 полных геномов SARS-CoV-2 с датой забора биоматериала с 01.01.2020 по 31.12.2023. Отбирали только те геномные последовательности, которые соответствовали заданным критериям качества. Отобранные геномы выравнивали на референсную последовательность NC_045512.2 с помощью средства NextClade и затем транслировали в аминокислотные последовательности. С помощью специализированного скрипта, написанного на языке Python, подсчитывали количество аминокислотных изменений по сравнению с референсом.
Результаты
Разработка и создание платформы агрегации вирусных геномов России VGARus
В 2021 г. на базе ЦНИИЭ в соответствии с постановлением Правительства РФ была разработана и создана платформа VGARus (Virus Genome Aggregator of Russia; дата регистрации 06.07.2023, № 2023622263). Ключевыми задачами этой платформы являются сбор данных о вирусных геномах, централизованный анализ генетического разнообразия и временнóй динамики выявленных вариантов SARS-CoV-2 в России. Фактически был создан научный консорциум, включающий учреждения Роспотребнадзора, Министерства здравоохранения Российской Федерации, различные научные институты и другие организации.
В настоящее время членами консорциума являются более 150 организаций, многие из которых активно проводят обширное геномное секвенирование SARS-CoV-2 и загружают полученные последовательности в базу данных VGARus для дальнейшего анализа. Кроме того, в проекте участвуют Республика Армения и Республика Беларусь, что позволяет отслеживать изменчивость патогенов в соседних странах с активными транспортными связями.
Процесс мониторинга изменчивости вирусных геномов включает следующие шаги (рис. 1):
- секвенирующая лаборатория получает биологический материал из диагностических лабораторий, включая те, что находятся при стационарах. Качество этих образцов предварительно оценивается, обычно с помощью ПЦР-анализа, для определения вирусной нагрузки и пригодности образца к секвенированию (NGS);
- лаборатория, предоставившая биологический материал, должна внести соответствующие метаданные в платформу VGARus (информация о поле, возрасте, статусе вакцинации пациента, дате забора биологического материала, регионе сбора и т.д.);
- специализированная лаборатория выполняет необходимую подготовку образцов и последующее секвенирование вирусных геномов;
- проводится первичный биоинформатический анализ, который включает контроль качества данных, сборку генома (обычно путём выравнивания на референсный геном) и проверку валидности последовательностей (оценка степени покрытости генома);
- загруженная геномная информация проходит валидацию и обрабатывается с использованием программ «Pangolin» [19, 20] для полных геномов и «V-TRACE» (разработка ЦНИИЭ) для результатов фрагментного секвенирования в автоматическом режиме. Образцы, для которых собранные геномы не проходят проверку качества, помечаются на платформе как невалидные. После этого в диагностическую лабораторию отправляется соответствующее уведомление.

Рис. 1. Этапы процесса мониторинга изменчивости вирусных геномов.
Fig. 1. The stages of the process of monitoring the variability of viral genomes.
Все геномные последовательности SARS-CoV-2 в стране, полученные в рамках рутинного эпидемиологического мониторинга, регистрируются в базе данных VGARus. Система поддерживает как ручной способ загрузки, так и загрузку с помощью специализированных API, что позволяет добавлять большое количество последовательностей. Каждый образец в системе содержит не только нуклеотидную последовательность, но и технические данные. При регистрации в базе данных образцу автоматически присваивается внутренний идентификатор, после чего последовательность генома SARS-CoV-2 добавляется в поле информации об образце. Помимо описанных ранее, техническая информация также включает в себя данные об организациях, участвующих в сборе образцов и лабораторной обработке, даты получения образца, регистрации материала в системе и загрузки последовательности.
S-белок SARS-CoV-2 играет ключевую роль в идентификации варианта вируса. Это связано с его ролью в проникновении вириона в клетки хозяина и высокой астотой мутаций/вариабельностью последовательности [21]. Однако попытка установить вариант вируса исключительно на основе мутаций в гене S-белка может привести к неполным или противоречивым результатам [22]. Учитывая сложность вирусной эволюции, при которой отдельные мутации могут по-разному влиять на функции вируса, а их комбинация может приводить к изменению общего эффекта, биоинформатики ЦНИИЭ разработали алгоритм V-TRACE для решения этой проблемы. Алгоритм идентифицирует мутации в гене S-белка SARS-CoV-2, после чего происходит оценка меры правдоподобия принадлежности исследуемой последовательности к различным линиям вируса.
Временнáя динамика и эволюционные траектории вариантов SARS-CoV-2
Платформа VGARus представляет собой ценный ресурс для отслеживания динамики развития пандемии COVID-19 в России и исследования её особенностей. В частности, систематически собираемая информация о последовательностях позволяет исследовать геномное разнообразие вируса.
В 2020 г. наблюдалось богатое разнообразие различных линий SARS-CoV-2 [23]. Они не демонстрировали значительных преимуществ по сравнению друг с другом, из-за чего ни один из вариантов не становился доминирующим. В декабре 2020 г., примерно через год после начала распространения нового коронавируса по всему миру, власти Великобритании сообщили Всемирной организации здравоохранения об обнаружении новой линии SARS-CoV-2, названной VOC-202012/01. Она имела многочисленные мутации в своём геноме и изначально была названа «британской», однако позже была переименована в Alpha, чтобы избежать наименования вариантов по странам. Среди мутаций, обнаруженных в гене S-белка, наиболее важными оказались N501Y, P681H и Δ69–70 [24, 25]. Эти мутации влияли на способность вируса инфицировать клетки и уходить от иммунного ответа хозяина и, как следствие, позволяли ему более эффективно распространяться. Этот вариант был обнаружен в России в конце 2020 г. и сохранялся в начале 2021 г., что совпало с резким увеличением числа случаев заболевания.
Вскоре после этого был идентифицирован вариант Beta, но он имел намного меньшее распространение, чем Alpha. Летом 2021 г. появился вариант Delta, быстро ставший доминирующим, что совпало со значительным ростом заболеваемости и уровня госпитализаций [26]. После периода относительно благоприятной эпидемиологической обстановки в декабре 2021 г. появился вариант Omicron (рис. 2), что привело к заметному увеличению числа случаев заболевания в России. Однако заболеваемость так же быстро снизилась.

Рис. 2. Диаграмма частот встречаемости значимых вариантов SARS-CoV-2 в России с мая 2020 г. по март 2024 г.
Fig. 2. A diagram illustrating the occurrence of significant SARS-CoV-2 variants of concern in the Russian Federation throughout 2020 until the March of 2024.
Несмотря на малое количество выявляемых случаев COVID-19 весной 2022 г., появление субвариантов Omicron BA.4 и BA.5 привело к росту заболеваемости, который продолжался до конца октября (рис. 3). В конце 2022 г. и начале 2023 г. появились высококонтагиозные варианты, такие как BQ.1*. Подобные смены доминирующих линий хорошо иллюстрируют постоянно меняющийся и сложный характер эволюции SARS-CoV-2. Примечательно, что в начале 2023 г. в популяцию вируса вернулись модифицированные версии ранее существовавших линий, в частности, Omicron BA.2, представленный в виде рекомбинантных форм XBB*. В ноябре 2023 г. в нескольких странах, в том числе в России, начал быстро распространяться вариант коронавируса BA.2.86, неофициально названный Pirola. Он был примечателен большим числом накопленных в геноме изменений по сравнению с более ранними линиями и к концу 2023 г. стал преобладающей линией вируса, а в начале 2024 г. его сублиния JN.1 почти полностью доминировала в большинстве стран мира.

Рис. 3. Диаграмма частот встречаемости различных сублиний варианта Omicron с января 2022 г. по март 2024 г.
Рост распространённости сублинии BA.2 к концу 2023 г. объясняется появлением варианта BA.2.86.
Fig. 3. Chart showing the frequency of occurrence of various Omicron sublineages from early 2022 to March of 2024.
The rise in the prevalence of the BA.2 sublineage by the end of 2023 is attributed to the BA.2.86 variant.
Описанная выше изменчивость патогена подчёркивает важность постоянного эпидемиологического мониторинга и секвенирования геномов вируса для своевременного выявления новых вариантов или изменений в структуре вирусной популяции. Оперативная идентификация таких изменений способна помочь при разработке стратегий общественного здравоохранения и при контроле распространения этих вариантов.
Сравнительный анализ рис. 2 и 3 показал, что существует тенденция, при которой каждая новая значимая линия вируса при занятии доминирующего положения достигала порогового значения 50% общего размера популяции приблизительно за 1,5–3,0 мес. Одновременно с этим период доминирования отдельной линии составлял от 3 мес до 1 года.
Затем мы попытались объяснить динамику заболеваемости COVID-19 в России и изучить возможность прогнозирования скорости распространения конкретной сублинии вируса на основе данных о последовательностях SARS-CoV-2. Наша основная гипотеза заключается в том, что специфические мутации в геноме вируса существенно влияют на уровень заболеваемости. Однако зарегистрированный уровень заболеваемости, несомненно, зависит и от других критических факторов, таких как уровень иммунизации населения, объёмы проведения ПЦР-тестирований и сезонные факторы, точный вклад которых трудно оценить. Поэтому эти факторы не были использованы для анализа.
Период с мая 2020 г. по декабрь 2023 г. был разделён на интервалы по 21 день. Исследуемый временной промежуток был ограничен декабрём 2023 г., что позволяет детально изучить тенденции, сложившиеся на тот момент. Однако последующее появление варианта BA.2.86 показало, что прогноз будущих трендов может быть чрезвычайно сложным. С учетом ключевой роли, которую играют миссенс-мутации в скорости передачи вируса, были проведены выравнивание нуклеотидных последовательностей на референсный геном и их трансляция в аминокислотные последовательности с помощью NextClade [27]. В качестве метрики вирусной изменчивости было выбрано количество аминокислотных изменений по сравнению с референсной последовательностью. Например, быстрый рост числа часто встречающихся изменений может указывать на активный мутационный процесс или на завоз новой линии в исследуемый регион. Чем выше скорость мутаций вируса, тем больше вероятность того, что некоторое подмножество приобретённых мутаций сможет повлиять на свойства вируса, например, на его трансмиссивность.
Аминокислотные изменения по сравнению с референсной последовательностью с мая 2020 г. по декабрь 2023 г. представлены черной линией на рис. 4. Для каждого интервала отображены только мутации с частотой не менее 50%. Такой подход позволяет оценивать мутации, которые либо существенно влияют на адаптивность вирусного варианта, либо могут наследоваться вместе с такими мутациями. На графике явно выделяются три различных интервала времени, в которые происходило увеличение числа частых мутаций в геноме SARS-CoV-2. Эти моменты соответствуют июню 2021 г., февралю 2022 г. и январю 2023 г. соответственно. Каждый из этих интервалов совпадает с широким распространением в России вариантов Delta, Omicron и XBB соответственно.

Рис. 4. Аминокислотные изменения по сравнению с референсной последовательностью с мая 2020 г. по декабрь 2023 г.
1 — число аминокислотных замен относительно референсного варианта (NC_045512.2) с частотой 50% и выше; 2 — число изменений множества мутаций с частотой не менее 50% по сравнению с предыдущим периодом; 3 — временнáя динамика случаев заболевания в России согласно данным ВОЗ.
Fig. 4. Changes in amino acid sequences compared to the reference sequence from May 2020 to December 2023.
1 — number of amino acid substitutions relative to the reference variant (NC_045512.2) with a frequency of 50% or higher; 2 — number of changes in the set of mutations with a frequency of at least 50% compared to the previous period; 3 — temporal dynamics of COVID-19 cases in Russia according to WHO data.
Одним из потенциальных ограничений описанного выше метода является то, что он не может эффективно отразить динамику изменений, когда новые и старые доминирующие линии имеют разные характерные мутации, но их абсолютное количество различается незначительно. Простой подсчёт мутаций в таком случае приведёт к ошибочному выводу о том, что генетической эволюции не происходит. Чтобы решить эту проблему, был проведён анализ качественного состава мутаций.
Наборы аминокислотных изменений с частотой более 50% были рассмотрены как отдельные математические множества для каждого временнóго интервала. Различия между ними для смежных временны́х интервалов были оценены и использованы для измерения генетической изменчивости вируса. В этом случае как появление, так и исчезновение мутации с частотой не менее 50% по сравнению с предыдущим периодом считалось изменением (рис. 4, 2). Эта вспомогательная стратегия продемонстрировала свою надёжность. Благодаря её применению мы наблюдали качественные изменения в наборе частых мутаций популяции, вызванных переходом от линии BA.1 к линии BA.2. При этом колебания в абсолютном числе распространённых мутаций оказались минимальными. Обе описанные выше совокупности данных согласуются с данными Всемирной организации здравоохранения о заболеваемости SARS-CoV-2 в России (рис. 4, 3).
Увеличению заболеваемости часто предшествуют значительные изменения в геноме патогена, как это было летом 2021 г. — появление варианта Delta, в декабре 2021 г. — Omicron (BA.1/BA.2), в июле 2022 г. — Omicron (BA.5), в начале 2023 г. — XBB (рис. 5). Однако сезонные факторы также играют важную роль.

Рис. 5. Динамика частотного распределения аминокислотных мутаций.
Линии показывают тенденции, связанные с изменениями доминирующей линии вируса. Каждая точка соответствует мутации, представленной на временнóй шкале, её положение указывает на частоту встречаемости на момент сбора образцов. Показаны основные линии, приводившие к изменениям частот мутаций. Пунктирными линиями выделены локальные тренды.
Fig. 5. Dynamics of the frequency distribution of amino acid mutations.
The lines indicate trends associated with changes in the dominant virus lineage. Each point represents a mutation plotted on the timeline, with its position indicating the frequency of occurrence at the time of sample collection. The major lineages leading to changes in mutation frequencies are shown. Local trends are highlighted with dashed lines.
Нами исследована динамика частот мутаций в геноме SARS-CoV-2 в России (рис. 5). Было отмечено, что при распространении новой доминантной линии в указанные выше моменты времени частота характерных для них мутаций демонстрировала быстрый S-образный рост. Когда новая линия начинала доминировать в стране, частота мутаций, характерных для заменяемой линии, снижалась по аналогичной S-образной траектории. Помимо общей картины, на графике распределения частот мутаций можно наблюдать локальные тренды.
Распределение частот мутаций также позволяет увидеть два ярко выраженных тренда в правой части графика. Более точный анализ мутаций и последовательностей, составляющих эти тренды, показывает, что обе группы линий включают XBB.1.9.1, FL.24, FL.1.5.1, XBB.1.16, XBB.1.16.11 и XBB.1.16.17. Верхний тренд, расположенный в области высоких частот (70–90%) и обозначенный пунктирной линией, формируется мутациями, общими для всех этих линий, такими как S:G252V и ORF1b:S959P. В то же время в области низких частот пунктирной линией обозначен нижний тренд, сформированный мутациями, обнаруженными в подгруппах тех же линий, например, S:E180V, представленная только в XBB.1.16, XBB.1.16.11 и XBB.1.16.17, и ORF1a:G1819S, обнаруженная в остальных линиях.
Изучение динамики распространения этих линий со временем может потенциально помочь прогнозировать эволюцию SARS-CoV-2. По этой причине динамика частоты для нескольких обсуждаемых сублиний с июня по декабрь 2023 г. представлена на рис. 6. Линии XBB.1.9.1 и XBB.1.16 были намеренно исключены из этого анализа в силу убывающего или статичного характера тренда их частот в рассматриваемый период. Первая линия значительно снизила свою распространённость почти до нуля, в то время как вторая не изменила существенно свою частоту, оставаясь в пределах 6–16%. Следовательно, они не рассматриваются как будущие потенциально доминирующие линии.

Рис. 6. Распространённость SARS-CoV-2 с июня по середину декабря 2023 г.
Fig. 6. Prevalence of SARS-CoV-2 from June 2023 through mid-December 2023.
Однако после этого периода линия BA.2.86* (Pirola) стала доминирующей вскоре после XBB, что не могло быть предсказано по данным, полученным к середине декабря 2023 г., когда эта линия была найдена лишь в нескольких образцах. Рисунок 6 также показывает, что большинство линий, ранее считавшихся потенциально доминирующими, хотя и становятся более распространёнными, достигают частоты лишь около 16% за несколько месяцев. Между тем, согласно более ранним наблюдениям в этом исследовании, доминирующая линия обычно достигает 50% частоты в течение 1,5–3,0 мес с момента появления.
Эти данные позволяют предположить, что будущая потенциально доминирующая линия должна иметь определённую минимальную скорость распространения; в противном случае она, вероятно, будет вытеснена другими. Такая гипотеза согласуется с периодической природой смены линий. Кроме того, такие события значительно усложняют прогнозирование хода пандемии, особенно в части определения доминирующей линии в ближайшем будущем и её влияния на здоровье населения. Наконец, быстрое появление и распространение совершенно нового варианта может сделать все предыдущие прогнозы неактуальными.
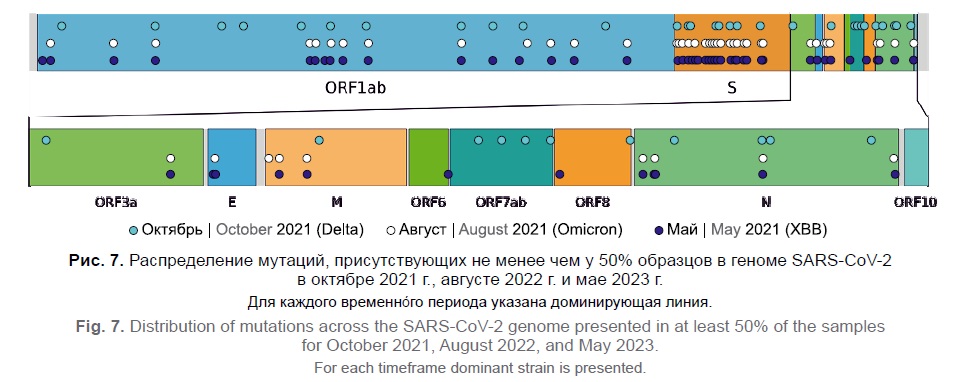
Также были получены данные о распределении мутаций в геноме. На рис. 7 обозначены мутации, достигшие частоты не менее 50% в рассматриваемые моменты времени. Многие из них обнаружены в гене S-белка, что согласуется с результатами других исследовательских групп [28]. Можно отметить, что каждая новая доминирующая линия привносит всё меньше новых мутаций в этот ген. Однозначно интерпретировать это наблюдение в настоящее время затруднительно, однако это может указывать на степень родства между линиями или на то, что структура гена приближается к оптимальной, максимизирующей аффинность к рецептору ACE2 человека.

Рис. 7. Распределение мутаций, присутствующих не менее чем у 50% образцов в геноме SARS-CoV-2 в октябре 2021 г., августе 2022 г. и мае 2023 г.
Для каждого временнÓго периода указана доминирующая линия.
Fig. 7. Distribution of mutations across the SARS-CoV-2 genome presented in at least 50% of the samples for October 2021, August 2022, and May 2023.
For each timeframe dominant strain is presented.
Обсуждение
Разработка российской платформы агрегирования вирусных геномов VGARus стала важным аспектом в борьбе с пандемией COVID-19 в стране. Эта база данных содержит более 320 000 последовательностей геномов SARS-CoV-2, из них около 200 000 полных геномов. Она помогает выполнять множество важных функций, таких как идентификация новых вариантов вируса, создание эффективных диагностических инструментов и формирование политики в области общественного здравоохранения [12–15]. VGARus значительно способствовала пониманию пространственной и временнóй динамики пандемии COVID-19. Предоставляя подробную информацию о времени и месте взятия каждого геномного образца, платформа позволяет визуализировать распределение конкретных вирусных вариантов в России и их эволюцию во времени. Это детальное знание даёт важное преимущество в предсказании эпидемиологических тенденций в ближайшие месяцы. Например, с использованием VGARus мы точно предсказали рост заболеваемости COVID-19 летом 2022 г. и в начале 2023 г., связанный с появлением субвариантов Omicron BA.5 и XBB соответственно. Эти прогностические возможности позволяют органам здравоохранения оперативно реагировать на эпидемиологическую ситуацию.
В целом представленные выше результаты о частотах встречаемости различных вариантов SARS-CoV-2 на территории России согласуются с данными других исследований в разных странах. Например, B. Xiao и соавт. использовали данные платформы GISAID [29]. Ими были рассмотрены последовательности, полученные в период с января 2020 г. по 20 мая 2023 г. в США, Великобритании, Индии, ЮАР, Бразилии и России. За этот период платформой GISAID было собрано около 79 000 геномов из России.
Общая картина эволюции и распространения вируса в разных странах схожа, однако имеются существенные региональные отличия. Например, в США и Великобритании пиковая доля варианта Alpha составляла около 65% и 100% соответственно. При этом в России оценка его пиковой распространённости составила около 40% по данным зарубежного исследования и 20% в текущей работе. Это различие может объясняться более широкой географией нашего исследования, нежели проведённого только по данным GISAID.
Вариант Delta попал в Россию примерно в тот же период, что и в другие рассмотренные страны, — в апреле–мае 2021 г. Исключение составила Индия, где этот вариант появился в марте 2021 г., а также Бразилия, где распространение штамма началось лишь в июле 2021 г. Как и в большинстве других изученных стран, в России практически не обнаруживался вариант Gamma, который при этом был доминирующим в Бразилии и регистрировался в США с пиковой частотой около 10%.
Частотная динамика сублиний BA.1 и BA.2 практически совпадает у России и всех исследованных стран, за исключением Индии, где BA.1 не стал доминирующим (менее 40%), а BA.2 распространился раньше и оставался доминирующим почти 8 мес. Уникальной для России и Индии ситуацией оказалось практически полное отсутствие варианта BA.4, который в остальных странах встречался с пиковой частотой 20–60%.
Представленное сравнение демонстрирует, что отдельные варианты вируса могут начинать своё распространение в разных регионах планеты с разницей в несколько месяцев, а время их максимальной распространённости может заметно варьировать. При этом данные о частотной динамике вариантов SARS-CoV-2 согласуются с выводами о сроках их распространения и доминирования, сделанными в данном исследовании.
В этом исследовании нами также были продемонстрированы основные возможности платформы VGARus, представлена динамика разнообразия вариантов и мутаций SARS-CoV-2 в России, а также подробно рассмотрен сценарий, предшествовавший смене доминирующей линии в декабре 2023 г. Результаты этой части исследования выявили определённые ограничения в предсказании доминирующей линии в ближайшем будущем. Эти ограничения, вызванные быстрым появлением новых линий, впоследствии становящихся доминирующими, представляют собой существенную проблему для имеющихся инструментов прогнозирования [30, 31]. Тем не менее мы не теряем надежды на то, что в будущем появятся подходы, которые будут эффективно учитывать и этот фактор. Несмотря на это, данная работа продемонстрировала некоторые общие тенденции в течении пандемии, среди которых важнейшее значение имеет периодический характер смены доминирующего варианта.
Помимо своей роли в отслеживании течения эпидемий, VGARus имеет практическое значение, такое как создание и тестирование диагностических тестов. Так, исследователи ЦНИИЭ регулярно используют данные VGARus для проверки эффективности праймеров и ДНК-зондов, используемых в тестовых наборах. Таким образом, платформа дает информацию для разработки и переработки олигонуклеотидов для диагностических тест-систем, информацию о циркулирующих в нашей стране вариантах, о вариабельности последовательности патогена в области «посадки» праймеров и зондов [32].
Возможности платформы VGARus теперь выходят за рамки исследований SARS-CoV-2. Платформа расширяется для предоставления данных о других патогенах, включая вирусы гепатита, гриппа, ветряной оспы, кори и многие другие. Такая возможность работы с несколькими патогенами станет ценным инструментом для вирусологов и специалистов по инфекционным заболеваниям, позволит им осуществлять более качественный мониторинг распространения различных болезней и своевременно принимать меры по защите общественного здравоохранения.
В целом VGARus является значительным достижением совместных усилий многочисленных научных институтов Роспотребнадзора и других ведомств [12]. Её внедрение расширило наше понимание SARS-CoV-2 и внесло вклад в изучение и борьбу с пандемией COVID-19. VGARus подчёркивает критическую важность эпидемиологического мониторинга в борьбе со вспышками инфекционных заболеваний и значимость совместных усилий в решении глобальных кризисов здравоохранения.
1 Novel 2019 Coronavirus Genome.
URL: https://virological.org/t/novel-2019-coronavirus-genome/319
Об авторах
Иван Андреевич Котов
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора; Московский физико-технический институт (национальный исследовательский университет)
Email: samurnihs@gmail.com
ORCID iD: 0000-0003-2416-5689
м.н.с. лаб. геномных исследований ЦНИИ Эпидемиологии Роспотребнадзора, аспирант МФТИ (НИУ)
Россия, Москва; ДолгопрудныйМатвей Рашидович Аглетдинов
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора; Московский физико-технический институт (национальный исследовательский университет)
Email: m.agletdinov2018@gmail.com
ORCID iD: 0000-0003-2249-7196
биоинформатик лаб. геномных исследований ЦНИИ Эпидемиологии Роспотребнадзора, аспирант МФТИ (НИУ)
Россия, Москва; ДолгопрудныйГерман Викторович Роев
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора; Московский физико-технический институт (национальный исследовательский университет)
Email: roevherman@gmail.com
ORCID iD: 0000-0002-2353-5222
биоинформатик лаб. геномных исследований ЦНИИ Эпидемиологии Роспотребнадзора, аспирант МФТИ (НИУ)
Россия, Москва; ДолгопрудныйЕкатерина Владимировна Пимкина
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: tsibina@cmd.su
ORCID iD: 0000-0002-0591-3525
м.н.с. научной группы геномных технологий лаб. геномных исследований
Россия, МоскваМаксим Игоревич Надтока
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: nadtoka@cmd.su
ORCID iD: 0009-0002-3217-0963
н.с. научной группы геномных технологий лаб. геномных исследований
Россия, МоскваАрина Валерьевна Пересадина
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: Peresadina@cmd.su
ORCID iD: 0009-0002-4981-6716
биоинформатик лаб. геномных исследований
Россия, МоскваАнна Юрьевна Бухарина
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: bukharina@cmd.su
ORCID iD: 0000-0002-6892-3595
м.н.с. научной группы геномных технологий лаб. геномных исследований
Россия, МоскваДмитрий Васильевич Светличный
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: svetlichnyi@cmd.su
ORCID iD: 0009-0008-6864-2807
м.н.с. научной группы геномных технологий лаб. геномных исследований
Россия, МоскваСергей Евгеньевич Гончаров
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: Goncharov@cmd.su
ORCID iD: 0009-0000-2842-5818
лаборант-исследователь научной группы геномных технологий лаб. геномных исследований
Россия, МоскваАнастасия Владимировна Выходцева
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: vihodceva@cmd.su
ORCID iD: 0009-0005-1911-9620
технолог научной группы геномных технологий лаб. геномных исследований
Россия, МоскваНадежда Ивановна Борисова
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: borisova@cmd.su
ORCID iD: 0000-0002-9672-0648
м.н.с. научной группы геномных технологий лаб. геномных исследований
Россия, МоскваВладислав Геннадиевич Лысенков
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: vladislav.lysenkov@gmail.com
ORCID iD: 0000-0002-1468-1631
биоинформатик лаб. геномных исследований
Россия, МоскваМихаил Дамирович Чанышев
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: Chanyshev@cmd.su
ORCID iD: 0000-0002-6943-2915
к.б.н., с.н.с. лаб. геномных исследований
Россия, МоскваДавид Накам оглы Агабалаев
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: Agabalaev@cmd.su
ORCID iD: 0000-0001-8125-355X
биоинформатик лаб. геномных исследований
Россия, МоскваВалерия Владимировна Саенко
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: valeriia.kaptelova@gmail.com
ORCID iD: 0000-0003-0952-0830
н.с. научной группы геномных технологий лаб. геномных исследований
Россия, МоскваАнна Сергеевна Черкашина
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: cherkashina@pcr.ms
ORCID iD: 0000-0001-7970-7495
к.х.н., рук. научной группы генной инженерии и биотехнологии
Россия, МоскваТатьяна Анатольевна Семененко
Национальный исследовательский центр эпидемиологии и микробиологии имени почётного академика Н.Ф. Гамалеи
Email: meddy@inbox.ru
ORCID iD: 0000-0002-6686-9011
д.м.н., профессор, научный консультант отдела эпидемиологии
Россия, МоскваДмитрий Васильевич Дубоделов
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: dubodelov@cmd.su
ORCID iD: 0000-0003-3093-5731
к.м.н., с.н.с. лаб. вирусных гепатитов
Россия, МоскваКамиль Фаридович Хафизов
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Автор, ответственный за переписку.
Email: khafizov@cmd.su
ORCID iD: 0000-0001-5524-0296
к.б.н., зав. лаб. геномных исследований
Россия, МоскваВасилий Геннадьевич Акимкин
Центральный научно-исследовательский институт эпидемиологии Роспотребнадзора
Email: vgakimkin@yandex.ru
ORCID iD: 0000-0003-4228-9044
д.м.н., профессор, академик РАН, директор
Россия, МоскваСписок литературы
- Barzon L., Lavezzo E., Militello V., et al. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 2011;12(11):7861–84. DOI: https://doi.org/10.3390/ijms12117861
- Quer J., Colomer-Castell S., Campos C., et al. Next-generation sequencing for confronting virus pandemics. Viruses. 2022;14(3):600. DOI: https://doi.org/10.3390/v14030600
- Capobianchi M.R., Giombini E., Rozera G. Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013;19(1):15–22. DOI: https://doi.org/10.1111/1469-0691.12056
- Singh D.D. Next-generation sequencing technologies as emergent tools and their challenges in viral diagnostic. Biomed. Res. 2018;29(8):1637–44. DOI: https://doi.org/10.4066/biomedicalresearch.29-18-362
- Mokili J.L., Rohwer F., Dutilh B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012;2(1): 63–77. DOI: https://doi.org/10.1016/j.coviro.2011.12.004
- Akermi S., Jayant S., Ghosh A., et al. Viroinformatics for Viral diseases: tools and databases. In: Translational Bioinformatics in Healthcare and Medicine. Elsevier;2021:171–82.
- Olson R.D., Assaf R., Brettin T., et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic. Acids. Res. 2023; 51(D1):D678–89. DOI: https://doi.org/10.1093/nar/gkac1003
- Shu Y., McCauley J. GISAID: Global initiative on sharing all influenza data — from vision to reality. Euro. Surveill. 2017;22(13):30494. DOI: https://doi.org/10.2807/1560-7917.ES.2017.22.13.30494
- Hill V., Ruis C., Bajaj S., et al. Progress and challenges in virus genomic epidemiology. Trends Parasitol. 2021;37(12):1038–49. DOI: https://doi.org/10.1016/j.pt.2021.08.007
- Giovanetti M., Slavov S.N., Fonseca V., et al. Genomic epidemiology of the SARS-CoV-2 epidemic in Brazil. Nat. Microbiol. 2022;7(9):1490–500. DOI: https://doi.org/10.1038/s41564-022-01191-z
- Klink G.V., Safina K.R., Nabieva E., et al. The rise and spread of the SARS-CoV-2 AY.122 lineage in Russia. Virus Evol. 2022;8(1):veac017. DOI: https://doi.org/10.1093/ve/veac017
- Akimkin V., Semenenko T.A., Ugleva S.V., et al. COVID-19 epidemic process and evolution of SARS-CoV-2 genetic variants in the Russian Federation. Microbiol. Res. 2024;15(1):213–24.
- Акимкин В.Г., Попова А.Ю., Плоскирева А.А. и др. COVID-19: эволюция пандемии в России. Сообщение I: проявления эпидемического процесса COVID-19. Журнал микробиологии, эпидемиологии и иммунобиологии. 2022; 99(3):269–286. Akimkin V.G., Popova A.Yu., Ploskireva A.A., et al. COVID-19: the evolution of the pandemic in Russia. Report I: manifestations of the COVID-19 epidemic process. Journal of microbiology, epidemiology and immunobiology. 2022;99(3):269–286. doi: 10.36233/0372-9311-276
- Акимкин В.Г., Попова А.Ю., Хафизов К.Ф. и др. COVID-19: эволюция пандемии в России. Сообщение II: динамика циркуляции геновариантов вируса SARS-CoV-2. Журнал микробиологии, эпидемиологии и иммунобиологии. 2022; 99(4):381–396. Akimkin V.G., Popova A.Yu., Khafizov K.F., et al. COVID-19: evolution of the pandemic in Russia. Report II: dynamics of the circulation of SARS-CoV-2 genetic variants. Journal of microbiology, epidemiology and immunobiology. 2022;99(4):381–396. doi: 10.36233/0372-9311-295
- Акимкин В.Г., Семененко Т.А., Хафизов К.Ф. и др. Стратегия геномного эпидемиологического надзора. Проблемы и перспективы. Журнал микробиологии, эпидемиологии и иммунобиологии. 2024;101(2):163–172. Akimkin V.G., Semenenko T.A., Khafizov K.F., et al. Genomic surveillance strategy. Problems and perspectives. Journal of microbiology, epidemiology and immunobiology. 2024;101(2):163–172. doi: 10.36233/0372-9311-507
- Hill V., Githinji G., Vogels C.B.F., et al. Toward a global virus genomic surveillance network. Cell Host Microbe. 2023;31(6): 861–73. DOI: https://doi.org/10.1016/j.chom.2023.03.003
- Lu R., Zhao X., Li J., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–74. DOI: https://doi.org/10.1016/S0140-6736(20)30251-8
- Corman V.M., Landt O., Kaiser M., et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25(3):2000045. DOI: https://doi.org/10.2807/1560-7917.ES.2020.25.3.2000045
- Rambaut A., Holmes E.C., O'Toole Á., et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5(11):1403–7. DOI: https://doi.org/10.1038/s41564-020-0770-5
- O'Toole Á., Scher E., Underwood A., et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus. Evol. 2021;7(2):veab064. DOI: https://doi.org/10.1093/ve/veab064
- Lan J., Ge J., Yu J., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–20. DOI: https://doi.org/10.1038/s41586-020-2180-5
- O'Toole Á., Pybus O.G., Abram M.E., et al. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genomics. 2022;23(1):121. DOI: https://doi.org/10.1186/s12864-022-08358-2
- Komissarov A.B., Safina K.R., Garushyants S.K., et al. Genomic epidemiology of the early stages of the SARS-CoV-2 outbreak in Russia. Nat. Commun. 2021;12(1):649. DOI: https://doi.org/10.1038/s41467-020-20880-z
- Liu Y., Liu J., Plante K.S., et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature. 2022;602(7896):294–9. DOI: https://doi.org/10.1038/s41586-021-04245-0
- Lista M.J., Winstone H., Wilson H.D., et al. The P681H mutation in the spike glycoprotein of the Alpha variant of SARS-CoV-2 escapes IFITM restriction and is necessary for type I Interferon resistance. J. Virol. 2022;96(23):e0125022. DOI: https://doi.org/10.1128/jvi.01250-22
- Борисова Н.И., Котов И.А., Колесников А.А. и др. Мониторинг распространения вариантов SARS-CoV-2 (Coronaviridae: Coronavirinae: Betacoronavirus; Sarbecovirus) на территории московского региона с помощью таргетного высокопроизводительного секвенирования. Вопросы вирусологии. 2021;66(4):269–78. Borisova N.I., Kotov I.A., Kolesnikov A.A., et al. Monitoring the spread of the SARS-CoV-2 (Coronaviridae: Coronavirinae: Betacoronavirus; Sarbecovirus) variants in the Moscow region using targeted high-throughput sequencing. Problems of Virology. 2021; 66(4):269–78. DOI: https://doi.org/10.36233/0507-4088-72 EDN: https://elibrary.ru/qdsujp
- Aksamentov I., Roemer C., Hodcroft E., Neher R. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021;6(67):3773. DOI: https://doi.org/10.21105/joss.03773
- Kumar R., Srivastava Y., Muthuramalingam P., et al. Understanding mutations in human SARS-CoV-2 spike glycoprotein: a systematic review & meta-analysis. Viruses. 2023;15(4):856. DOI: https://doi.org/10.3390/v15040856
- Xiao B., Wu L., Sun Q., et al. Dynamic analysis of SARS-CoV-2 evolution based on different countries. Gene. 2024;916:148426. DOI: https://doi.org/10.1016/j.gene.2024.148426
- Fernandes Q., Inchakalody V.P., Merhi M., et al. Emerging COVID-19 variants and their impact on SARS-CoV-2 diagnosis, therapeutics and vaccines. Ann. Med. 2022;54(1):524–40. DOI: https://doi.org/10.1080/07853890.2022.2031274
- Raghwani J., du Plessis L., McCrone J.T., et al. Genomic epidemiology of early SARS-CoV-2 transmission dynamics, Gujarat, India. Emerg. Infect. Dis. 2022;28(4):751–8. DOI: https://doi.org/10.3201/eid2804.212053
- Kotov I., Saenko V., Borisova N., et al. Effective approaches to study the genetic variability of SARS-CoV-2. Viruses. 2022;14(9):1855. DOI: https://doi.org/10.3390/v14091855
Дополнительные файлы

